
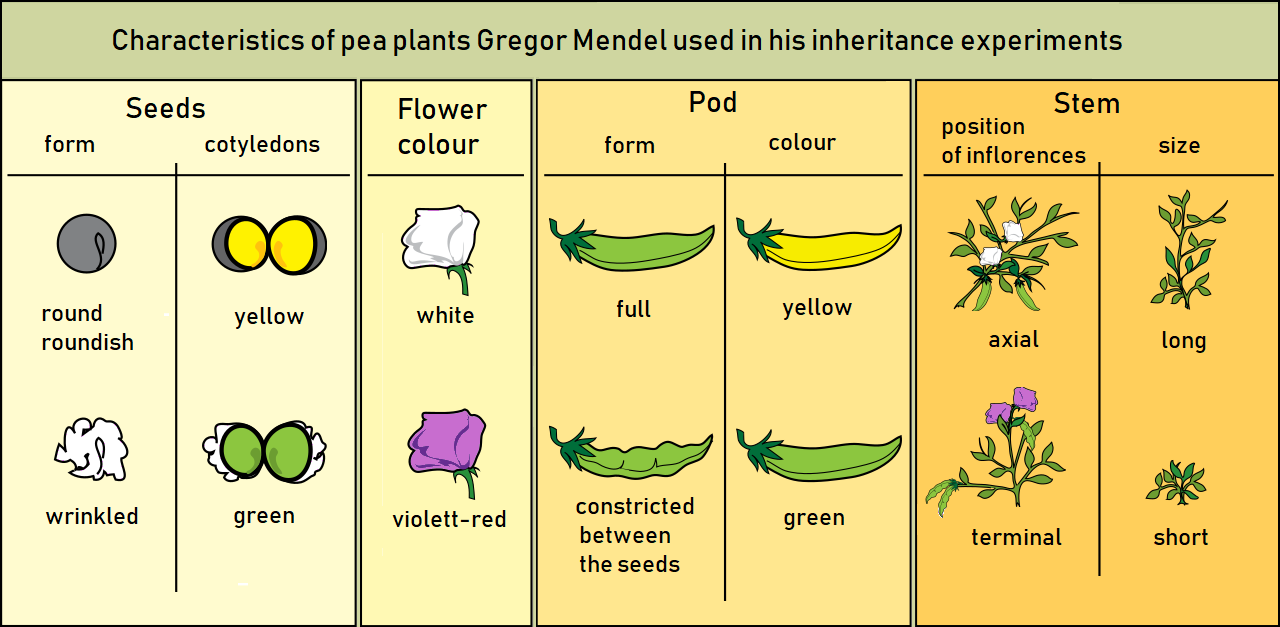
Infectious disease genotyping has become a mainstream topic due to the COVID-19 pandemic, but finding variants and reporting their significance is as old as the field of genetics itself. The famous pea-plant experiments that Gregor Mendel carried out in the mid 1800, introduced the idea of traits – some inheritable characteristic, observable differences, that can change from generation to generation (Figure 1).
{kind=link}
Fast-forward to today, where we have infectious disease genotyping (the process of determining the DNA sequence) of viruses and bacteria to find mutations that change transmissibility, infectiousness and resistance patterns from one generation to the next. Instead of comparing a yellow pod to a green pod, or a long stem and a short stem (Figure 1), we talk about the infectiousness of SARS-CoV-2 Delta and Omicron, or HIV-1 viruses that are resistant or not resistant to a specific antiretroviral drug.
One of the challenges for reporting these characteristics about transmissibility, infectiousness and resistance, is that each mutation has to be associated with the characteristic – does it cause a small or large change in transmissibility? and how many times has it been observed? are some of the questions scientists have to ask while conducting infectious disease genotyping research.
Although variant characterisation of different infectious diseases can be a massive undertaking, the resulting databases of mutations and characteristics are essential for infectious disease genotyping and reporting. Software and platforms, like Exatype, use these databases to translate observed DNA sequence mutations to interpretable characteristics. Therefore, while one of the most important, and challenging. steps in DNA sequencing analysis is to find real mutations within the large amounts of noisy DNA data, this mutation-to-characteristic translation is what makes the result easily interpretable and meaningful.
Our infectious disease genotyping solutions for HIV (Exatype HIV NGS, Exatype HIV Sanger and Exatype HIV NGS Proviral) incorporate the global standard database, Stanford HIVDB, for resistance reporting and interpretation, while our SARS-CoV-2 solutions (Exatype SARS-CoV-2 and Exatype SARS-CoV-2 HPR) use Nextclade and Pangolin for lineage/clade typing, i.e. reporting variants such as Delta and Omicron. While the reported characteristics are very different for HIV and SARS-CoV-2, the principle of translating a mutation to characteristic is the same. It is for this reason that the Exatype platform is not limited to reporting only one type of infectious disease genotyping characteristic of selected viruses or organisms, but is designed such that any mutation-to-characteristic translation, for any organism, can be added to the platform.
We are excited to add more solutions in 2022 - if you’re looking for a DNA sequencing analysis partner for your infectious disease genotyping assay, please get in touch!